(m) Synonym: Apo-B/E-Rezeptor
Zellrezeptor, der eine zentrale Rolle für den LDL-Katabolismus und damit bei der Elimination von →Cholesterin aus dem Plasma spielt. Der LDL-Rezeptor ist ein kettenförmiges Protein, das aus 839 →Aminosäuren besteht und in 5 Domänen unterteilt ist: Liganden bindende, Epidermal-Growth-Factor-(EGF-)homologe, zuckerhaltige, Membran durchquerende und zytoplasmatische Domäne. Die Liganden bindende Domäne besteht aus 7 Sequenzen, die mit Cystein angereichert sind und mittels elektrostatischer Kräfte die arginin- und lysinreichen Abschnitte des Apo B100 binden Abb. 27 .
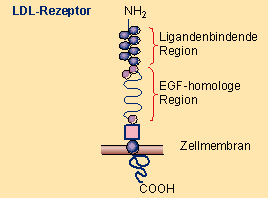
Abb. 27 Struktur des LDL-Rezeptors.
Zellen tragen etwa 40 000 LDL-Rezeptoren auf ihrer Oberfläche in clathrinbeschichteten Grübchen (Coated Pits). Zellen, die Cholesterin zur Synthese von Gallensäuren bzw. Hormonen (Leber bzw. Nebennieren, Gonaden) benötigen, sind dichter mit LDL-Rezeptoren besetzt. Hauptaufgabe der LDL-Rezeptoren ist, eine konstante Zulieferung von Cholesterin an die Zellen zu gewährleisten. LDL-Rezeptoren binden →LDL und internalisieren sie, die entstandenen Vesikel bilden intrazelluläre Endophagosomen. Bei einem pH-Wert von 5 werden LDL dort von den LDL-Rezeptoren abgelöst, die zur erneuten Verwendung wieder zur Zelloberfläche transportiert werden. Die LDL in den Phagolysosomen werden abgebaut, die darin enthaltenen Cholesterinester werden zu freiem Cholesterin hydrolysiert, und die Proteine der LDL zu Aminosäuren metabolisiert. Das freie Cholesterin regelt die intrazelluläre Cholesterinhomöostase vorwiegend mittels dreier Mechanismen:
- Unterdrückung der zelleigenen Cholesterinsynthese durch Inhibierung der →HMG-CoA-Reduktase.
- Verminderung der LDL-Rezeptor-Synthese.
- Gesteigerte Wiederveresterung von freiem Cholesterin mit Oleat durch das Enzym Acyl-CoA-Cholesterin-Acyltransferase (ACAT). Überschüssiges entstandenes Cholesteryloleat wird in Vakuolen gespeichert (Schaumzelle).
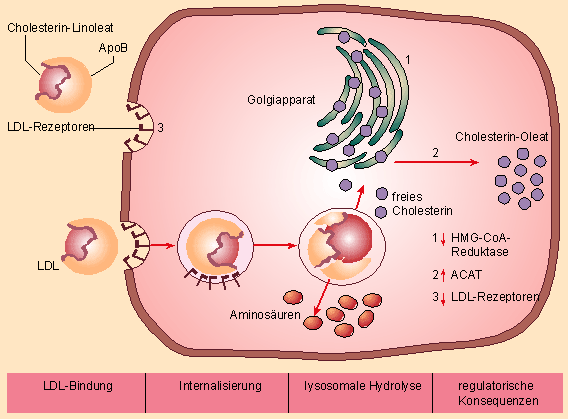
Abb. 28 Regulationsmechanismen des intrazellulären Cholesterinspiegels. Das via LDL-Rezeptor in die Zelle eingeschleuste Cholesterin wird zu freiem Cholesterin metabolisiert, das nun die zelleigene Synthese drosselt (1), die Speicherung in Vakuolen steigert (2) und als wichtigste Konsequenz die Ausbildung der LDL-Rezeptoren auf der Zelloberfläche vermindert (3).
Defekte des LDL-Rezeptors können zu verminderter Bindung und Internalisierung von Plasma-LDL führen und sind Ursache der verschiedenen Formen der familiären →Hypercholesterinämie. Kommen die Defekte heterozygot vor, haben die Betroffenen nur ca. 50 % funktionsfähige LDL-Rezeptoren; das →Gesamtcholesterin liegt dann zwischen 200 bis ca. 500 mg/dl. Bei homozygoten LDL-Rezeptor-Defekten kann das Gesamtcholesterin über 1000 mg/dl betragen. Bis jetzt sind mehr als 100 LDL-Rezeptor-Defekte bekannt. Betroffen sind eine oder mehrere Eigenschaften des Rezeptors wie Rezeptorsynthese (Klasse 1), Transport und Stoffwechsel von Vorläuferproteinen (Klasse 2), Ligandenbindung (Klasse 3), Rezeptorgruppierung und Internalisierung (Klasse 4) und Rezeptor-„Recycling“ und -Degradation (Klasse 5). Die Häufigkeit der heterozygoten Defekte beträgt in den bisher untersuchten Populationen 1:500. Sollte die familiäre →Hypercholesterinämie mit der gleichen Frequenz auch in weiteren Regionen der Erde vorkommen, dürfte es sich hierbei um die häufigste, durch eine einzige Genmutation verursachte Erkrankung des Menschen handeln. Die Entdeckung des LDL-Rezeptors und die Erforschung seiner Bedeutung für den Cholesterinmetabolismus führten zur Verleihung des Nobelpreises an Michael S. Brown und Joseph Goldstein im Jahre 1985.